
11.1.8: Models of Modern Human Distribution
- Last updated
- Save as PDF
- Page ID
- 136453
There has been much debate in anthropological circles concerning the origin of modern humans and their relationship with other hominin populations. Three competing models have been developed and seek to explain the fossil evidence and what it indicates for modeling human origins.
The first model, the Out-of-Africa Hypothesis, states that modern humans originated in Africa, replacing archaic populations found elsewhere in the Old World. Theorists including Christopher Stringer (1996) argue that each archaic population comprised a separate species, making interbreeding between populations impossible. Admixture resulting from gene flow would not have been possible according to this model.
The second, called the Multiregional Continuity Hypothesis, states that modern Homo sapiens are directly derived from Homo erectus and evolved in place after Homo erectus left Africa and populated areas in Asia and Europe. Milford Wolpoff argues that interbreeding between regions and across regional boundaries contributed to gene flow that maintained Homo sapiens as a single species throughout the Old World, despite regional variation.
The third model, dubbed the Assimilation Hypothesis, draws from the strengths of both previous models, attempting to recognize some of the evidence that was not previously addressed and blending the fossil and DNA evidence together into one cohesive view. In this model, modern humans originated in Africa, spreading outward into Asia and Europe and interbreeding with more archaic forms they encountered along the way. For example, while the Out-of-Africa model argues that interbreeding would have been impossible, many fossils have been found with what appear to be a mixture of archaic and more modern traits, suggesting interbreeding between populations, such as Neanderthals and modern humans. DNA evidence increasingly also suggests that, while limited, interbreeding between modern Homo sapiens and Neanderthals or modern Homo sapiens and Denisovans occured in at least three instances. While this is more interbreeding than allowed under the Out-of-Africa Hypothesis, it is considerably less than modeled in the Multiregional Continuity Hypothesis. The Assimilation Hypothesis, argued by Eric Trinkaus (2006, 2007) and others, represents an attempt to incorporate all lines of evidence, although new research will tell whether it can capture the full complexity revealed in the next generation of hominin studies, such as that revealed by ancient DNA.
SPECIAL TOPIC: ANCIENT DNA
Robyn Humphries, MSc., University of Cape Town
Ancient DNA has provided us with new insights into our evolutionary history that cannot be garnered from the fossil record. It has also assisted with the discovery of the new hominin species the Denisovans, for which little fossil evidence is available. It has helped us better understand the evolution of Neanderthals, Denisovans, and modern humans. Through genomic data and the use of population genetics, we have been able to make some inferences about Neanderthal and Denisovan population structure and relationships within these populations as well as between different groups of hominins. It has also helped to answer some very important questions about what happened when modern humans migrated out of Africa and encountered these European/Asian hominins. Two theories dominated the debate regarding the evolution of modern humans: the multiregional theory and the Out-of-Africa theory. Though it was clear—based on a plethora of evidence—that modern humans evolved in Africa, what happened when our ancestors migrated out of Africa was still questioned. Ancient DNA (aDNA) helped answer this question, indicating that modern humans interacted with other archaic hominins such as Neanderthals and Denisovans. We will discuss all the above in this section.
Sequencing Ancient Genomes
The first successful sequencing of aDNA from an archaic hominin took place in 1997 with the sequencing of mitochondrial DNA (mtDNA) from the Neanderthal-type specimen from Feldhofer Cave. Sequencing of a portion of the mitochondrial genome provided molecular evidence that Neanderthals belonged in a clade separate from modern humans and that they were four times more different from modern humans than modern humans were from each other based on mtDNA data. mtDNA is ideal for sequencing from fossil material because of the abundance of mtDNA when compared to nuclear DNA.
Sequencing of nuclear DNA would not occur until more than ten years later. The first nuclear genomic sequence representing Neanderthals was produced by sequencing three individuals and using their sequences to create a composite draft Neanderthal genome in 2010. The first high-coverage sequence of a single Neanderthal was that of a female Neanderthal who lived in Siberia, which was published in 2014, followed by another high-coverage sequence from a female Neanderthal whose remains were found in the Vidja cave in Croatia, which was published in 2017. are produced when the genome has been sequenced multiple times. This is to ensure that the sequences obtained are a true reflection of the genomic sequence and not due to errors that occur during the process of sequencing. If you have many sequences from the same region and there is one sequence that has a slight difference while the other copies are all the same, it is easier to identify the variant as an error.
Collecting and Sequencing aDNA
Ancient DNA can be collected from many different sources including soft tissue such as skin and muscle, hair, paleo feces, soils, and sediments. However, in the case of ancient hominins, they are often collected from bone and teeth. When collecting aDNA, usually around 100 mg to 500 mg of bone powder needs to be collected. Because extraction of aDNA requires destruction of part of the bone, and the morphology of the skeletal element might be informative, care needs to be taken when deciding which part of the bone is sampled. It is advised that multiple samples be taken so that sequencing is repeated to show reproducibility of results. Contamination is an important consideration when it comes to sequencing aDNA; thus, it is best that samples that are used had minimal handling before extraction of DNA.
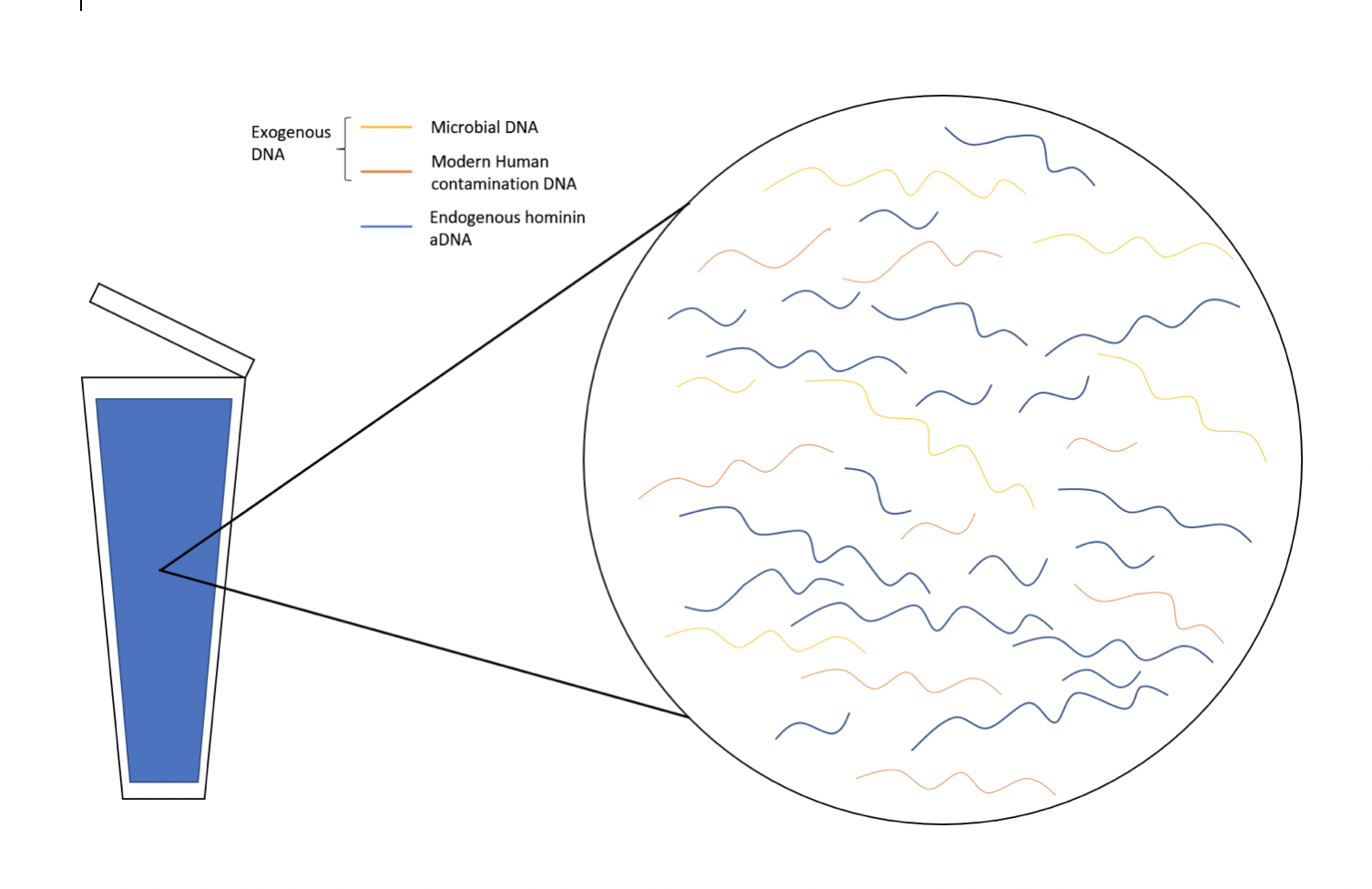 Figure \(\PageIndex{1}\): An illustration of the different types of DNA you may find after DNA extraction is performed on bone or other samples.
Figure \(\PageIndex{1}\): An illustration of the different types of DNA you may find after DNA extraction is performed on bone or other samples.It has taken a lot of time and much trial and error to sequence these ancient genomes because of the fragility of DNA. When sequencing ancient DNA, it is important to consider that aDNA sequences are usually short due to degradation, there are very few copies of the endogenous aDNA. Endogenous aDNA is the DNA that comes from the bone and was present in the tissue before decomposition of the body and before the introduction of DNA from other sources, such as microbes or contamination from modern humans, which is known as exogenous DNA (Figure 11.12).
There are also modifications that occur to aDNA that are a result of chemical reactions known as deamination. Deamination results in Cytosine (C) to Thymine (T) conversions, which are mostly at the 5’ end ()of the DNA fragment. This in turn results in Guanine (G) to Adenine (A) substitutions on the 3’ end ()of the DNA fragment. Thus, there are sequence changes in aDNA that might not reflect the original hominin sequence. These changes can be helpful when differentiating between aDNA and modern human DNA contamination. The environment in which the DNA is preserved also plays a significant role. DNA preserves well in cold conditions such as permafrost, which extends the lifespan of DNA significantly. aDNA has also been recovered from material found in drier environments under special conditions. Factors such as water percolation, salinity, pH, and microbial growth all affect the preservation of aDNA.
In extraction of DNA from modern samples where DNA is still intact, the DNA strands are usually long and this is ideal for sequencing. However, aDNA samples are often composed of small fragments of DNA, usually 100 bp to 300 bp long. Initially this posed a big problem with usual PCR procedures used to sequence DNA. This changed with the advent of high throughput sequencing, which has revolutionized sequencing the genomes of ancient hominins. High throughput sequencing allows for the parallel sequencing of many fragments of DNA in one reaction. It also doesn’t require any knowledge of the target sequence. Thus, we can sequence as much of the available aDNA as possible. Because the high throughput sequencing method does not discriminate between endogenous aDNA from hominins and contamination from modern humans and microbial DNA, it is important to either ensure that there is as little contamination as possible or create methods that allow for differentiation between modern human sequences and ancient hominin sequences. Both methods have been used when sequencing hominin aDNA.
The Discovery of the Denisovans
The Denisovans are named after the cave in which they were discovered, the Denisovan Cave in the Altai Region of Siberia. Denisovans were initially identified as a distinct group based on analysis of mtDNA sequences indicating that they had haplotypes outside the range of variation of modern humans and Neanderthals. A haplotype is a set of genetic variants located on a single stretch of the genome. This unique combination of variants on a stretch of the genome can be used to differentiate groups who will have different combinations of variants. Some haplotypes may be more similar to one another. The more similar two haplotypes are, the more closely related they are. Dubbed lineage X, the mtDNA sequence showed that Denisovans diverged from modern humans and Neanderthals at around 1 million years ago (mya). The subsequent high-coverage sequence of a Denisovan 3 nuclear genome showed that Denisovans are a sister group to Neanderthals and thus more closely related than indicated by the mtDNA data.
The mtDNA and nuclear DNA provided conflicting data regarding the relationships between Denisovans and Neanderthals. Because mtDNA and nuclear DNA have different patterns of inheritance, they can paint different pictures about the relationships between two groups when used to construct phylogenies. The Denisovans are thought to have a mtDNA sequence that is derived from an ancient hominin group that hybridized with Denisovans and introduced the mtDNA sequence.
Sequences are also available for three other Denisovans, Denisovan 2, 4, and 8. aDNA sequences have been used to estimate the ages of the Denisovans. Using a combination of usual dating methods (such as radio carbon dating and uranium dating) as well as genetic data, it has been determined that Denisovans occupied the Denisovan cave from around 195 kya to 52 kya to 76 kya. DNA can assist with dating because younger sequences will have accumulated more sequence changes from the putative common ancestral sequence than older samples. This is because younger sequences would have had more time over which changes to the DNA sequence through mutation could occur. Thus, it is possible to conclude based on sequence data that Denisovan 2 is 54.2 kya to 99.4 kya older than Denisovan 3 and 20.6 kya to 37.7 kya older than Denisovan 8. Molecular data indicates that Neanderthals and Denisovans separated between 381 kya and 473 kya and that the branch leading to Denisovans and modern humans diverged around 800 kya. Denisovans are also more closely related to another set of fossils found in the cave Sima de los Huesos dated to 480 kya. Thus, the split between Neanderthals and Denisovans must have occurred before 480 kya.
What Can We Learn about Population Structure of the Neanderthals and Denisovans from aDNA
Ancient DNA has helped us understand the demographics of Neanderthals and Denisovans and make inferences about population size and history. The genomic data from Neanderthals indicates that their population was small toward the end of their existence. This is supported by three lines of evidence.
The first is by using coalescent methods. This is the process used to determine which population dynamics in the past are most likely to give rise to the genetic sequences we have, allowing us to use genetic sequences to estimate population genetic parameters in the past. It can be used to understand recombination, population subdivision, and variable population size.
The second indicator that Neanderthals and Denisovans had smaller population size is that these groups carried many deleterious genomic variants. Genomic variants are considered deleterious when they are found in protein-coding regions of the genome and the change in genomic sequence translates to a change in amino acid sequence of the protein. Changes in amino acid at a certain section in the protein could affect the functioning of the protein—these types of changes in genomic sequence are known as non-synonymous mutations. Synonymous mutations also occur in protein-coding regions of the genome, but the amino acid sequence does not change because of changes in the genomic sequence. Changes in amino acid and subsequent protein sequence can change the protein function and thus are more likely to be deleterious and weeded out by natural selection. The ratio of synonymous to can give you an indicator of whether there are more deleterious variants than expected. Denisovans and Neanderthals have a higher ratio of non-synonymous to synonymous mutations when compared to contemporary modern human populations. This is an indicator of a small population size, because if the population were larger, natural selection would have acted on these deleterious variants and weeded them out.
A third indicator of small population size is that the Neanderthals sequenced thus far have low levels of heterozygosity, a measure of how many genes within a genome are made up of more than one variant. Each individual has two copies of the same gene: one is inherited from their mother and the other from their father. Variations of the same gene are known as alleles, which are versions of the same gene with h different sequences. If the alleles inherited from both parents are the same, the individual is homozygous for that gene; if the alleles inherited are different, the individual is heterozygous for that gene. Heterozygosity is measured by looking at how many times you happen to find two different alleles within a certain stretch of DNA. When you find many regions on the genome with different alleles, there is a high level of heterozygosity. When you find very few positions where there are two different alleles, this results in a low level of heterozygosity.
Ancient Neanderthal genomes also revealed that there were consanguineous relations between Neanderthals. One Neanderthal female is thought to be the offspring of relations between either half-siblings, an uncle/aunt and niece/nephew, or a grandfather/grandmother and grandson/granddaughter. This was determined by looking at the stretches of homozygosity in her genome that were longer than expected and could not be explained by small population size alone.
Denisovans also had low levels of heterozygosity indicating a smaller population size. However, there is no indication yet of inbreeding among the Denisovans, as none of the individuals sequenced thus far show long stretches of homozygosity. Thus, both Denisovans and Neanderthals had small populations size and low levels of genetic diversity when compared to modern humans.
How Sequencing Archaic Genomes Can Help Understand Our Own Unique Evolutionary Trajectory as Modern Humans
Not only did the sequencing of archaic genomes allow us to learn more about Neanderthals and Denisovans, it gave us important insights into our own evolution. Previously the human genome could only be compared to our closest living relatives, the great apes, which helped us identify unique derived genomic changes that occurred in humans since our split from the last common ancestor between chimps and humans. Neanderthal and Denisovan genomes provided another set of comparative samples that could help us identify changes that were unique to modern humans occurring after our split from the last common ancestor with Neanderthals/Denisovans. We now have an opportunity to identify genetic variants that may have contributed to our success as a species.
Hybridization between Hominin Groups
Ultimately aDNA provides us with great insight into interactions between modern humans migrating out of Africa and other hominins that evolved in Europe and Asia. There was speculation that hybridization occurred due to the intermediate morphology of some fossil remains. The following hypothesis was tested: if hybridization between modern humans and Neanderthals occurred, Neanderthals would have more shared genomic variants with some modern human populations than with others. If this was true, hybridization between Neanderthals and humans happened. This comparison showed that Neanderthals shared more genomic variants with Europeans and Asians than with the African individuals. This difference in relatedness was significant. This indicated that there had been hybridization between Neanderthals and modern humans.
From the genetic data, we know that different groups have different amounts of Neanderthal and Denisovan contributions. For example, Europeans have a smaller proportion of Neanderthal-derived genes than East Asians. Thus, there was more admixture into ancestral East Asian populations than into ancestral European populations. This is unexpected because Neanderthals fossils are mostly found in West Asia and Europe. Oceanians (Melanesians, Australian aborigines, and other Southeast Asian islanders) have a higher proportion of their DNA derived from Denisovans. These populations also have longer stretches of Denisovan DNA. Since DNA in chromosomes get exchanged and “break apart” between each generation (in the process known as genetic recombination), this implies that the admixture event between the Denisovan and human ancestors of these populations is more recent than the admixture events between Neanderthals and modern humans. Genomic recombination breaks down introgressed regions (inherited from different species or taxon) into smaller segments in every successive generation, thus longer stretches of introgressed DNA indicates that hybridization occurred more recently.
Initially, some researchers believed that populations outside of Africa had higher proportions of Neanderthal DNA due to population substructure, which existed in the ancestral population before the split between modern humans and Neanderthals. According to these researchers, Eurasian populations retained these ancient sequences by chance, through genetic drift. These would be shared derived genomic sequences between Neanderthals and modern humans outside of Africa. However, studies of the shared regions indicate that these genomic regions are most likely the result of introgression, which is the transfer of genetic information from one species to another because of hybridization between them and repeated backcrossing.
Divergence time is important for determining whether shared sequences are a result of introgression or more ancient substructure. Divergence time is a measure of how long two sequences have been changing independently. It is measured by looking at how many differences there are between the two sequences. The longer the two sequences have been changing independently, the more differences they will accumulate, which will result in a longer divergence time. By measuring the divergence time between the introgressed regions in modern human genomes and the Neanderthal sequences, researchers can calculate that the shared sequences are recent as well as date to when the two taxa made secondary contact. This is also well after the initial population split between modern humans and Neanderthals occurred. If they were shared derived genomic sequences, then we would expect longer divergence times between the introgressed Neanderthal genomic sequences in modern humans and the Neanderthal genome. Hybridization has occurred between hominins at different times over the last 100 kya as shown in Figure 11.14.
The Neanderthal and Denisovan genomes would provide definitive proof that there was interaction and interbreeding between humans, Neanderthals, and Denisovans around 44 to 55 kya, with data suggesting that there was an admixture event as far back as 100 kya. There has been gene flow from Neanderthals and Denisovans into modern human populations, between Neanderthals and Denisovans, and from modern humans into Neanderthals.
Because of the climate in Africa, it has been difficult or impossible due to fossilization to extract aDNA from African fossil remains. However, analysis of genomes of modern African populations indicate that there was admixture between modern humans and other hominins within Africa (Figure 11.13). Thus, hybridization is an important part of human evolution and has affected our evolution within and outside of Africa.
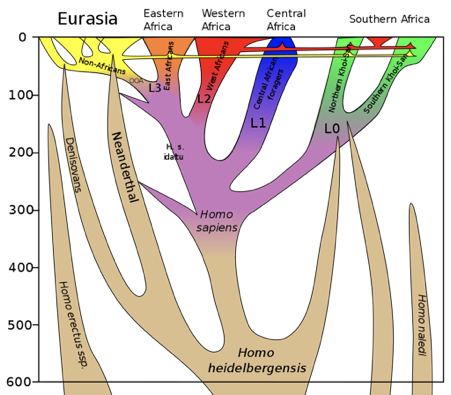 Figure \(\PageIndex{2}\): Phylogeny showing the relationship between modern humans and other hominins over the last 500 kya. This image is also depicting a number of hybridization events—for example, the genetic contributions that Neanderthals and Denisovans made to modern humans around 50 kya.
Figure \(\PageIndex{2}\): Phylogeny showing the relationship between modern humans and other hominins over the last 500 kya. This image is also depicting a number of hybridization events—for example, the genetic contributions that Neanderthals and Denisovans made to modern humans around 50 kya.The oldest modern human that has been sequenced Ust’-Ishim is from Europe and is dated to around 49 kya. He had a similar amount of Neanderthal-derived genes as modern humans from outside of Africa. Analysis of the genome indicated that the hybridization event resulting in the introgression occurred 50 ky before. The fact that the Ust’-Ishim modern human had longer tracts of Neanderthal-derived DNA than contemporary populations lends support to the idea that Neanderthal-derived DNA in modern humans is due to hybridization. Contemporary modern humans have shorter stretches of Neanderthal-derived genes because there has been a longer period over which the Neanderthal segments of DNA could be broken down by recombination.
Thus, there are multiple lines of evidence supporting hybridization between modern humans and Neanderthals/Denisovans. This includes shorter divergence times between introgressed regions in modern-human and Neanderthal sequences, older modern-human sequences having longer tracts of Neanderthal-derived genes and, as discussed below, the sequencing of confirmed hybrids.
Confirmed Fossil Hybrids
When discussing hybrids, there are some important terms to understand. A first-generation hybrid is called an F1 hybrid; it is the direct offspring of two lineages that have been evolving independently over an extended period. A second-generation hybrid (F2) would be the offspring of two F1 hybrids. A backcrossed individual is the result of an F1 or F2 hybrid mating with an individual from one of the parental populations. An example of a backcross would be when a Neanderthal-human hybrid produces offspring with a human; their offspring would be considered a first-generation backcrossed hybrid (B1). Sequencing of aDNA from fossil material has further confirmed that hybridization between different hominins has occurred, supporting the introgression data from recent populations.
The sequencing of Oase 1, a suspected hybrid based on skeletal morphology (what the fossil looked like), showed that it had a Neanderthal ancestor as recently as six to eight generations back. He would thus be considered a backcrossed individual. The recent sequencing of a 13-year-old Denisovan female showed that she was the F1 hybrid offspring of a Neanderthal mother (from whom she inherited Neanderthal mtDNA) and a Denisovan father. She was confirmed to be an F1 hybrid because approximately 50% of her genome was derived from a Neanderthal and 50% from a Denisovan.
These are only two examples of individuals who are confirmed hybrids. Many other remains show some indication of gene flow between hominins.
Neanderthal-and Denisovan-Derived DNA in Modern Genomes
There is variation in how much of the Neanderthal genome is represented in the modern human population. Individuals outside of Africa usually have 1% to 2 % of their genome derived from Neanderthals. Approximately 30% of the Neanderthal genome is represented in modern human genomes. Asian populations usually have a higher proportion of their genome derived from Neanderthals when compared to modern European populations. Additionally, the sequencing of the Denisovan genome indicates that they interacted with the ancestors of modern Oceanic populations. Thus, oceanic populations have around 5% to 6% of their genome derived from Denisovans. There is also evidence that different Denisovans populations may have contributed to Oceanians and East Asians. The available Denisovan sequences are more similar to the Denisovan introgressed genes found in East Asian populations.
Introgressed genes have signatures that allow us to identify them and differentiate them from parts of the genome that are not introgressed. Some of the things to look for when determining if a segment of the genome is introgressed include the following. First, how closely does the segment you are looking at match the Neanderthal/Denisovan sequence compared to contemporary modern human sequences from Africa? If the sequence is more similar to the Neanderthal sequence (i.e., it has less sequence differences from the Neanderthal than the African modern human), it is likely that it is derived from a Neanderthal). Second, what is the divergence time between the allele and the same allele in a Neanderthal? If it is shorter than the divergence time between humans and Neanderthal, then the gene is most likely introgressed. This is expected because if the divergence time is after the split between modern humans and Neanderthals, the most likely explanation for a shorter divergence time is introgression. An example of this can be seen in Figure 11.14. And, third and finally, you need to look at whether the allele is found at higher frequencies in populations outside of Africa.
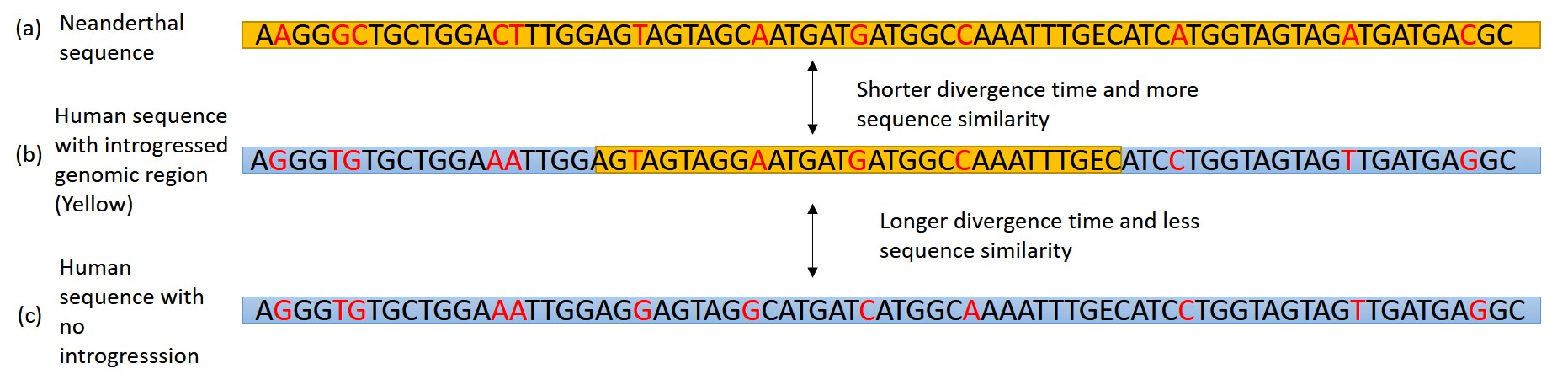 Figure \(\PageIndex{3}\): An illustration of a (b) introgressed region (in yellow) in a modern human genome and how it compares to the same segment in a (c) modern human with no introgression and an (a) Neanderthal sequence.
Figure \(\PageIndex{3}\): An illustration of a (b) introgressed region (in yellow) in a modern human genome and how it compares to the same segment in a (c) modern human with no introgression and an (a) Neanderthal sequence.What Can We Learn about the Process of Hybridization from Ancient DNA?
Ancient DNA has also allowed us to make certain inferences about the process of hybridization between modern humans and Neanderthals/Denisovans. From looking at the genomes of modern humans, we can see that there are regions of the genome with no Neanderthal and Denisovan genomic variants. These are known as Neanderthal or Denisovan introgression deserts. There are also overlaps between regions in the human genome that are Neanderthal and Denisovan deserts, which might indicate that there were genomic incompatibilities between modern humans and these groups, resulting in those genes being selected against on the modern human genome background. This resulted in strong negative selection against these genomic variants in subsequent generations of hybrids and backcrossed individuals.
We can also infer that hybridization may itself have been a barrier to gene flow because there is a significant reduction in introgression on the X chromosome compared to the other chromosomes. There is also a reduction of introgressed genes around genes that are disproportionately expressed in the testes when compared to other tissue groups. This could indicate that hybridization between modern humans and Neanderthals may have resulted in male hybrid infertility.
Hybridization and Modern Human Evolution
Hybridization provided adaptive advantage to modern humans migrating out of Africa by providing them with advantages in genetic variation. Neanderthals and Denisovans had spent hundreds of thousands of years adapting to the European and Asian environments and thus had genetic variants favorable for inhabiting those regions. Through hybridization, humans were able to acquire favorable genomic variants already selected for, and these variants could rapidly spread through the population. This allowed for faster adaptation because acquiring new variation through mutation alone is much slower and less likely to spread through the population. Some of the adaptive genes that were important include genes associated with immunity, adapting to new diets, adapting to new altitudes as well as genes involved in skin color and hair traits were introgressed. An excellent example of this would be a variant of the EPAS1 gene found at high frequencies in Tibetans, thought to be important for living at high altitudes. This variant of EPAS1 has been shown to be an introgressed gene from Denisovans.
The Future of Genetic Studies
We are continuing to learn how introgressed genes affect modern humans. Combining phenotypic and genetic information Neanderthal derived genes have been associated with diverse traits such as the skin’s sensitivity to the sun to excessive blood clotting by certain individuals. Interesting research has also shown that introgressed alleles might produce different gene expression profiles when compared to non-introgressed alleles. However, there is a lot of research that needs to be done to fully understand the effects of introgression on modern populations and how it might have assisted modern humans who migrated out of Africa.
It has also been possible to extract DNA from sediments. However, extractions from sediments will result in extraction of DNA from multiple organisms. To extract the hominin sequences, we will need to use the known sequence information. Known sequences will also assist with differentiating between which hominins are represented in the sediment. This could assist with identifying changes in populations across time. The availability of more Neanderthal and Denisovan samples will also help us understand which genetic changes were fixed and defined these populations.