
6.2: Human Variation in Biological Anthropology Today
- Last updated
- Save as PDF
- Page ID
- 130061
“Populations” Instead of “Races”
 Figure \(\PageIndex{1}\): Theodosius Dobzhansky, an important scientist who formulated the 20th-century “modern synthesis” reconciling Charles Darwin’s theory of evolution and Gregor Mendel’s ideas on heredity.
Figure \(\PageIndex{1}\): Theodosius Dobzhansky, an important scientist who formulated the 20th-century “modern synthesis” reconciling Charles Darwin’s theory of evolution and Gregor Mendel’s ideas on heredity.After 1950, replacing the concept of “race” as a unit of diversity was the “population.” This was outlined by those pioneering the “new physical anthropology,” such as Sherwood Washburn, Theodosius Dobzhansky, and Julian Huxley, who borrowed this way of framing human groups from contemporary population geneticists (Figure 13.10). “Races” were then defined simply as populations that differ in the frequency of some gene or genes. And, on the other hand, a “population” is a group of individuals potentially capable of or actually interbreeding due to shared geographic proximity, language, ethnicity, culture, and/or values. Put another way, a population is a local interbreeding group with reduced gene flow between themselves and other groups of humans. Members of the same population may be expected to share many genetic traits (and, as a result, many phenotypic traits that may or may not be visible outwardly).
 Figure \(\PageIndex{2}\): Julian Huxley (1942).
Figure \(\PageIndex{2}\): Julian Huxley (1942).Thinking of humans in terms of populations was part of Julian Huxley’s (1942) “Modern Synthesis”—so named because it helped to reconcile two fundamental principles about evolution that had not been made sense of together before (Figure 13.11). As discussed in Chapter 3, Gregor Mendel (1822‒1884) was able to show that inheritance was mediated by discrete particles (or genes) and not blended in the offspring. However, it was difficult for some 19th-century scientists to accept this model of genetic inheritance at the time because much of biological variation appeared to be continuous and not particulate (take skin color or height as examples). In the 1930s, it was demonstrated that traits could be polygenic and that multiple alleles could be responsible for any one phenotypic trait, thus producing the continuous variation in traits such as eye color that we see today. Thus, Huxley’s “Modern Synthesis” outlines not only how human populations are capable of exchanging genes at the microevolutionary level but also how multiple alleles for one trait (polygenic exchanges) can cause gradual macroevolutionary changes.
Human Variation Is Clinal/Continuous (Not Discrete)
Human diversity cannot be broken into discrete “races,” because most physical traits vary on a continuous or “clinal” basis. One obvious example of this is how human height does not only come in three values (“short,” “medium,” and “tall”) but instead varies across a spectrum of vertical heights achievable by humans all over the world. (However, this is with the only difference being the huge divergence in how factors like body size and traits such as skin color have been viewed and used sociopolitically as a way of separating people throughout history.) The need to shift from typological “race” categories to a more nuanced understanding of continuously variable populations was realized by anthropologists working in the 1960s and 1970s who shifted their focus toward the study of individual traits rather than the study of groups (populations, races). Systematic evaluations of global biological variation in humans only began then, when large numbers of genetic loci for large numbers of samples were sampled from human populations distributed worldwide. It was during the 1960s that “clines” in human genetic variation were first identified.
Frank B. Livingstone (1928‒2005) wrote: “There are no races, only clines” (1962). A cline is a gradation in the frequency of an allele/trait between populations living in different geographic regions. In order to study human traits that are clinally distributed, it is often required to perform genetic testing to uncover the true frequencies of an allele or trait across a certain geographic space. One easily visible example of a clinal distribution seen worldwide is the patterning of human variation in skin color. Whether in southern Asia, sub-Saharan Africa, or Australia, dark brown skin is found. Paler skin tones are found in higher-latitude populations such as those who have lived in areas like Europe, Siberia, and Alaska for millennia. Skin color is easily observable as a phenotypic trait exhibiting continuous variation.
A clinal distribution still derives from genetic inheritance, but clines often correspond to some gradually changing environmental factor. Clinal patterns arise when selective pressures in one geographic area differ from those in another as well as when people procreate and pass on genes together with their most immediate neighbors. There are several mechanisms, selective and neutral, that can lead to the clinal distribution of an allele or a biological trait. Natural selection is the mechanism that produced a global cline of skin color, whereby darker skin color protects equatorial populations from high amounts of UV radiation; there is a transition of lessening pigmentation in individuals that reside further and further away from the tropics (Jablonski 2004; Jablonski and Chaplin 2000) (Figure 13.12). The ability and inability to digest lactose (milk sugar) among different world communities varies according to differential practices and histories of milk and dairy product consumption (Gerbault et al. 2011; Ingram et al. 2009). Where malaria seems to be most prevalent as a disease stressor on human populations, a clinal gradient of increasing sickle cell anemia experience toward these regions has been studied extensively by genetic anthropologists (Luzzatto 2012). Sometimes culturally defined mate selection based on some observable trait can lead to clinal variation between populations as well.
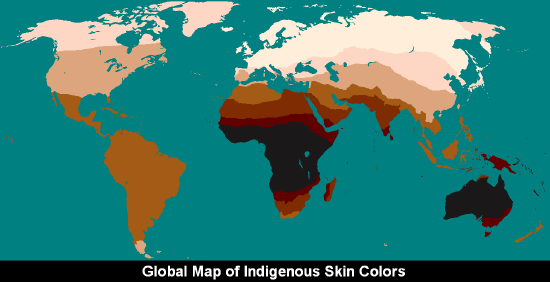 Figure \(\PageIndex{3}\): Global map of indigenous skin colors.
Figure \(\PageIndex{3}\): Global map of indigenous skin colors.Two neutral microevolutionary processes that may produce a cline in a human allele or trait are gene flow and genetic drift. The ways in which neutral processes can produce clinal distributions is seen clearly when looking at clinal maps for different blood groups in the human ABO blood group system (Figure 13.13). For instance, scientists have identified an east-to-west cline in the distribution of the blood type B allele across Eurasia. The frequency of B allele carriers decreases gradually westward when we compare the blood groups of East and Southeast Asian populations with those in Europe. This shows how populations residing nearer to one another are more likely to interbreed and share genetic material (i.e., undergo gene flow). We also see 90%‒100% of native South American individuals, as well as between 70%‒90% of Aboriginal Australian groups, carrying the O allele (Mourant et al. 1976). These high frequencies are likely due to random genetic drift and founder effects, in which population sizes were severely reduced by the earliest O allele-carrying individuals migrating into those areas. Over time, the O blood type has remained predominant.
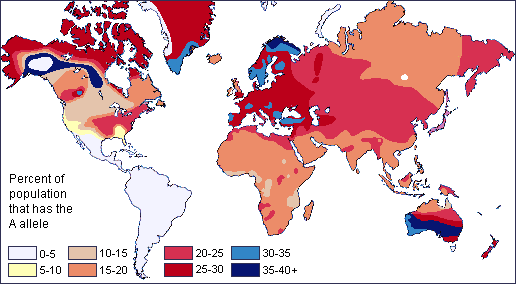.png?revision=1) Figure \(\PageIndex{4}\): Global distribution of blood group A.
Figure \(\PageIndex{4}\): Global distribution of blood group A.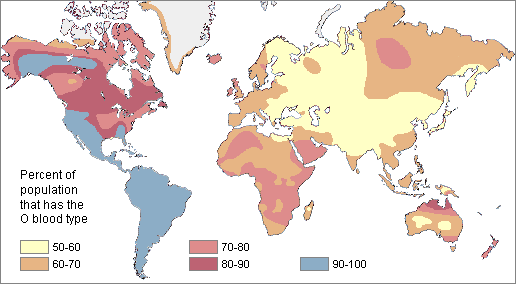 Figure \(\PageIndex{5}\): Global distribution of blood type B.Figure \(\PageIndex{6}\): Global distribution of blood type O.
Figure \(\PageIndex{5}\): Global distribution of blood type B.Figure \(\PageIndex{6}\): Global distribution of blood type O.The Apportionment of Human Variation: Genetic Diversity Is Greater Within-Group Than Between-Groups
One problem with race-based classifications is they relied on an erroneous idea that people within a typological category were more similar to each other than they were to people in other groups. In other words, “race” concepts were predicated on the notion that individuals with particular characteristics would share more similar genes with each other within a particular “race” and share less with individuals of other “races” possessing different traits and genetic makeups. However, since around 50 years ago, scientific studies have shown that the majority of human genetic differences worldwide exist within groups (or “races”) individually rather than between groups.
Richard Lewontin (1929‒) is a biologist and evolutionary geneticist who authored a paper evaluating where the total genetic variation in humans lies. This article, titled “The Apportionment of Human Diversity” (Lewontin 1972), addressed the following question: On average, how genetically similar are two randomly chosen people from the same group when compared to two randomly chosen people from different groups? Lewontin studied this problem by using genetic data. He obtained data for a large number of different human populations worldwide using 17 genetic markers (including alleles that code for various important enzymes and proteins, such as blood-group proteins). The statistical analysis he ran used a measure of human genetic differences in and among populations known as the fixation index (FST). Technically, FST can be defined as the proportion of total genetic variance within a subpopulation relative to the total genetic variance from an entire population. Therefore, FST values range from 0 to 1 (or, sometimes you will see this stated as a percentage between 0% and 100%). The closer the FST value of a population (e.g., the world’s population) approaches 1, the higher the degree of genetic differentiation among subpopulations relative to the overall population. In his paper, Lewontin (1972) identified that most of human genetic differences (85.4%) were found within local subpopulations (e.g., the Germans or Easter Islanders), whereas 8.3% were found between populations within continental human groups, and 6.3% were attributable to traditional “race” groups (e.g., “Caucasian” or “Amerind”). These findings have been important for scientifically rejecting the existence of biological races (Long and Kittles 2008).
In 2002, another landmark article by Noah Rosenberg and colleagues (2002) explored worldwide human genetic variation using an even-greater genetic data set. They used 377 highly variable markers in the human genome and sampled from 1,056 individuals representative of 52 populations. The markers chosen for study were not ones that code for any expressed genes. Because these regions of the human genome were made of unexpressed genes, we may understand these markers as neutrally derived (as opposed to selectively derived) as they do not code for functional advantages or disadvantages. These neutral genetic markers likely reflect an intricate combination of regional founder effects and population histories. Analyses of these neutral markers allowed scientists to identify that a majority of global genetic variance (93%‒95%) can be accounted for by within-population differences at the 377 genetic loci, while only a small proportion of genetic variance (3%‒5%) can be attributed to differences among major groups (Rosenberg et al. 2002). Like Lewontin’s (1972) findings, this lends support to the theory that distinct biological races do not exist, even though misguided concepts of race may still have real social and political consequences.
Biological Data Fit Isolation-By-Distance and Out-of-Africa Models
One further note is that while the world’s population may be genetically divided into “groups,” “subsets,” “clumps,” or “clusters” that reflect some degree of genetic similarity, it is more likely that these identifiable clusters reflect genetic or geographic distances—either with gene flow facilitated by proximity between populations or impeded by obstacles like oceans or environmentally challenging habitats (Rosenberg et al. 2005). Sometimes, inferred clusters using multiple genetic loci are interpreted by non-geneticists literally as “ancestral populations.” However, it would be wrong to assume from these genetic results that highly differentiated and “pure” ancestral groups ever existed. These groupings reflect differences that have arisen over time due to clinal patterning, genetic drift, and/or restricted or unrestricted gene flow (Weiss and Long 2009). The clusters identified by scientists are arbitrary and the parameters used to split up the global population into groups is subjective and dependent on the particular questions or distinctions being brought into focus (Relethford 2009).
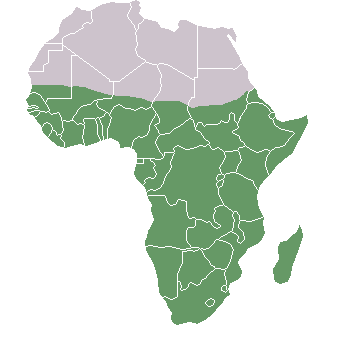 Figure \(\PageIndex{7}\): Sub-Saharan Africa (shaded dark/green).
Figure \(\PageIndex{7}\): Sub-Saharan Africa (shaded dark/green).Additionally, research on worldwide genetic diversity has shown that human variation decreases with increasing distance from sub-Saharan Africa, where there is evidence for this vast region being the geographical origin of anatomically modern humans ( Liu et al. 2006; Prugnolle et al. 2005) (Figure 13.14). Genetic differentiation decreases in human groups the further you sample data from relative to sub-Saharan Africa because of serial founder effects (Relethford 2004). Over the course of human colonization of the rest of the world outside Africa, populations broke away in expanding waves across continents into western Asia, then Europe and eastern Asia, followed by Oceania and the Americas. As a result, founder events occurred whereby genetic variation was lost, as the colonization of each new geographical region involved a smaller number of individuals moving from the original larger population to establish a new one (Relethford 2004). The most genetic variation is found across populations residing in different parts of sub-Saharan Africa, while other current populations in places like northern Europe and the southern tip of South America exhibit some of the least genetic differentiation relative to all global populations.
Besides fitting nicely into the , worldwide human genetic variation conforms to an , which predicts that genetic similarity between groups will decrease exponentially as the geographic distance between them increases. This is because of the greater and greater restrictions to gene flow presented by geographic distance, as well as cultural and linguistic differences that occur as a result of certain degrees of isolation. Since genetic data conform to isolation-by-distance and Out-of-Africa models, these findings support the abolishment of “race” groupings. This research demonstrates that human variation is continuous and cannot be differentiated into geographically discrete categories. There are no “inherent” or “innate” differences between human groups; instead, variation derives from some degree of natural selection, as well as neutral processes like population bottlenecking (Figure 13.15), random mutations in the DNA, genetic drift, and gene flow through between-mate interbreeding.
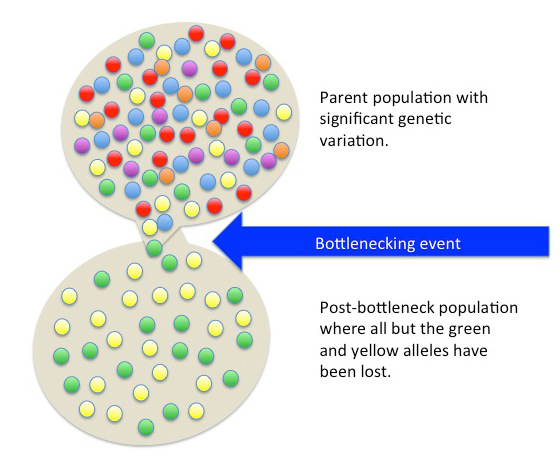 Figure \(\PageIndex{8}\): The founder effect is a change in a small population’s gene pool due to a limited number of individuals breaking away from a parent population.
Figure \(\PageIndex{8}\): The founder effect is a change in a small population’s gene pool due to a limited number of individuals breaking away from a parent population.Humans Have Higher Homogeneity Compared to Many Other Species
An important fact to bear in mind is that humans are 99.9% identical to one another. This means that the apportionments of human diversity discussed above only concern that tiny 0.1% of difference that exists between all humans globally. Compared to other mammalian species, including the other great apes, human diversity is remarkably lower. This may be surprising given that the worldwide human population has already exceeded seven billion, and, at least on the surface level, we appear to be quite phenotypically diverse. Molecular approaches to human and primate genetics tells us that external differences are merely superficial. For a proper appreciation of human diversity, we have to look at our closest relatives in the primate order and mammalian class. Compared to chimpanzees, gibbons, and even gray wolves and giant pandas, humans have remarkably low average genome-wide heterogeneity.
 Figure \(\PageIndex{9}\): Chimpanzee (Pan troglodytes).
Figure \(\PageIndex{9}\): Chimpanzee (Pan troglodytes).When we look at chimpanzee genetic diversity, it is fascinating that western, central, eastern, and Cameroonian chimpanzee groups have substantially more genetic diversity between them than large global samples of human DNA (Bowden et al. 2012) (Figure 13.16). This is surprising given that all of these chimpanzee groups live relatively near one another in Africa, while measurements of human genetic diversity have been conducted using samples from entirely different continents. First, geneticists suppose that this could reflect differential experiences of the founder effect between humans and chimpanzees. Because all non-African human populations descended from a small number of anatomically modern humans who left Africa, it would be expected that all groups descended from that smaller ancestral group would be similar genetically. Second, our species is really young, given that we have only existed on the planet for around 150,000 to 300,000 years. This gave humans little time for random genetic mutations to occur as genes get passed down through genetic interbreeding and meiosis. Chimpanzees, however, have inhabited different ecological niches, and less interbreeding has occurred between the four chimpanzee groups over the past six to eight million years compared to the amount of gene flow that occurred between worldwide human populations (Bowden et al. 2012).
Recent advances have now enabled the attainment of genetic samples from the larger family of great apes and the evaluation of genetic diversity among bonobos, orangutans, and gorillas alongside that of chimpanzees and humans (Prado-Martinez et al. 2013). Collecting such data and analyzing primate genetic diversity has been important not only to elucidate how different ecological, demographic, and climatic factors have shaped our evolution but also to inform upon conservation efforts and medical research. Genes that may code for genetic susceptibilities to tropical diseases that affect multiple primates can be studied through genome-wide methods. Species differences in the genomes associated with speech, behavior, or cognition could tell us more about how human individuals may be affected by genetically derived neurological or speech-related disorders and conditions (Prado-Martinez et al. 2013; Staes et al. 2017). In 2018, a great ape genomic study also reported genetic differences between chimpanzees and humans related to brain cell divisions (Kronenberg et al. 2018). From these results, it may be inferred that cognitive or behavioral variation between humans and the great apes might relate to an increased number of cortical neurons being formed during human brain development (Kronenberg et al. 2018). Comparative studies of human and nonhuman great ape genetic variation highlight the complex interactions of population histories, environmental changes, and natural selection between and within species. When viewed in the context of overall great ape diversity, we may reconsider how variable the human species is relatively and how unjustified previous “race” concepts really were.
Phenotypic Traits That Reflect Neutral Evolution
Most human traits are non-concordant. “” is a term used to describe how biological traits vary independent of each other—that is, they don’t get inherited in a correlative manner with other genetically controlled traits. For example, if you knew an individual had genes that coded for tall height, you would not be able to predict if they are lighter-skinned or have red hair. Depending on the trait being observed, different patterns of phenotypic variation may be found within and among groups worldwide. In this subsection, some phenotypic traits that reflect the aforementioned patterns of genetic variation will be discussed.
 Figure \(\PageIndex{10}\): Human skulls in Tana Toraja (Indonesia), common scenery in public graves.
Figure \(\PageIndex{10}\): Human skulls in Tana Toraja (Indonesia), common scenery in public graves.Looking beyond genetic variation briefly, recent studies have revisited biological anthropology’s earlier themes of externally observable traits, such as skull shape (Figure 13.17). In the last 20 or so years, anthropologists have evaluated the level to which human cranial shape diversity reflects the results from genetic markers, such as those used previously to fit against Out-of-Africa models (Relethford 2004) or those used in the apportionment of human diversity between and within groups (Lewontin 1972; Rosenberg et al. 2002). Using larger sample sizes of cranial data collected from thousands of skulls worldwide and a long list of cranial measurements, studies demonstrate a similar decrease in diversity with distance from Africa and show that a majority of cranial variation occurs within populations rather than between populations (Betti et al. 2009; Betti et al. 2010; Manica et al. 2007; Relethford 2001; von Cramon-Taubadel and Lycett 2008). The greatest cranial diversity is found among skulls of sub-Saharan African origin, while the least variation is found among populations inhabiting places like Tierra del Fuego at the southern tip of Argentina and Chile. While ancient and historical thinkers previously thought “race” categories could reasonably be determined based on skull dimensions, modern-day analyses using more informative sets of cranial traits simply show that migrations out of Africa and the relative distances between populations can explain a majority of worldwide cranial diversity (Betti et al. 2009).
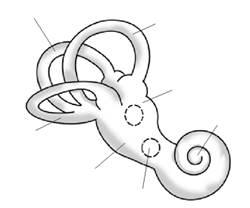 Figure \(\PageIndex{11}\): Diagram of the bony labyrinth in the inner ear.
Figure \(\PageIndex{11}\): Diagram of the bony labyrinth in the inner ear.This same patterning in phenotypic variation has even been found in studies examining shape variation of the pelvis (Betti et al. 2013; Betti et al. 2014), the teeth (Rathmann et al. 2017), and the human bony labyrinth of the ear (Ponce de León et al. 2018) (Figure 13.18). The skeletal morphology of these bones still varies worldwide, but a greater proportion of that variation can still be attributed to the ways in which human populations migrated across the world and exchanged genes with those closer to them rather than those further away. Human skeletal variation in these parts of the body is continuous and non-discrete. Given the important functions of the cranium and these other skeletal parts, we may infer that the genes that underpin their development have been relatively conserved by neutral evolutionary processes such as genetic drift and gene flow. It is also important to note that while some traits such as height, weight, cranial dimensions, and body composition are determined, in part, by genes, the underlying developmental processes behind these traits are underpinned by complex polygenic mechanisms that have led to the continuous spectrum of variation in such variables among modern-day human populations.
Phenotypic Traits That Reflect Natural Selection
Even though 99.9% of our DNA is the same between all humans worldwide, and many traits reflect neutral processes, there are parts of that remaining 0.1% of the human genome that code for individual and regional differences. Similarly to craniometric analyses that have been conducted in recent decades, human variation in skin color has also been reassessed using new methods and in light of greater knowledge of biological evolution.
New technologies allow scientists to use color photometry to sample and quantify the visible wavelength of skin color, in a way 19th- and 20th-century readers could not. In one report, it was found that 87.9% of global skin color variation can be attributed to genetic differences between groups, 3.2% to those among local populations within regions, and 8.9% within local populations (Relethford 2002). This apportionment differs significantly and is the reverse situation found in the distribution of genetic differences we see when we examine genetic markers such as blood type–related alleles. However, this pattern of human skin color worldwide is not surprising, given that we now understand that past selection has occurred for darker skin near the equator and lighter skin at higher latitudes (Jablonski 2004; Jablonski and Chaplin 2000). While most genetic diversity reflects neutral variation due to population migrations, geographic isolation, and restricted gene flow dynamics, some human genetic/phenotypic diversity is best explained as local adaptation to environmental conditions (i.e., selection). Given that skin color variation is atypical compared to other genetic markers and biological traits, this, in fact, goes against earlier “race” typologies. This is because recent studies ironically show how so much of genetic variation relates to neutral processes, while skin color does not. It follows that skin color cannot be viewed as useful in making inferences about other human traits.
On top of social implications, the quantification and interpretation of human variation has important medical and clinical applications (National Research Council Committee on Human Genome Diversity 1997). For instance, large-scale genomic studies sampling from human populations distributed worldwide have produced detailed knowledge on variation in disease resistance or susceptibility between and within populations. If we think about drug companies who develop medicines for African American patients particularly, the genetic diversity in predispositions to disease and/or good health is likely higher among people of African descent than these pharmaceutical businesses have taken into account. Through targeted sampling of various world groups, clinical geneticists may also identify genetic risk factors of certain common disorders such as chronic heart disease, asthma, diabetes, autoimmune diseases, and behavioral disorders. Having an understanding of population-specific biology is crucial in the development of therapies, medicines, and vaccinations, as not all treatments may be suitable for for every human, depending on their genotype. During diagnosis and treatment, it is important to have an evolutionary perspective on gene-environment relationships in patients. Typological concepts of “race” are not useful, given that most racial groups (whether self-identified or not) popularly recognized lack homogeneity and are, in fact, variable. Cystic fibrosis, for instance, occurs in all world populations but can often be underdiagnosed in populations of African ancestry because it is thought of as a “white” disease (Yudell et al. 2016).
 Figure \(\PageIndex{12}\): The Forensic Anthropology Lab at the National Museum of Natural History, Smithsonian Institute, Washington, D.C.
Figure \(\PageIndex{12}\): The Forensic Anthropology Lab at the National Museum of Natural History, Smithsonian Institute, Washington, D.C.Lastly, assignments of “race” to human remains is a common practice in forensic anthropology, especially in the United States and other worldwide contexts where bones are recovered and associated with criminal investigations (Figure 13.19). Forensic anthropologists have ascribed “race” or ancestry to sets of skeletons thanks to scientific research that has attempted to divide up different human groups into culturally constructed categories based on biologically “discrete” assortments (Sauer 1992). Rather than focusing on the neutral or selective causes of human biological variation, the concentration in forensic anthropology centers upon how probabilistic it may be to assign bones of certain dimensions to one of several identified racial categories. Forensic anthropologists do not agree with the typological “race” concepts of the past but, instead, root their racial categorization in methods of probability estimation (Sauer 1992). Based on many samples of skeletons from different world regions, statistical tests (such as discriminant function analysis) allow them to distinguish how likely certain skeletal dimensions may predict geographic ancestry.
It is important to remember that while it is possible to determine geographic origin (or ancestry) based on skull morphology, again, the amount of craniometric distinctiveness required to distinguish whether a cranium belongs to one group or another will make for arbitrary decisions (Relethford 2009). Individuals can vary in their skeletal dimensions by continental origin, country origin, regional origin, sex, age, environmental factors, and the time period in which they lived, making it difficult to assign individuals to particular categories in a completely meaningful way (Ousley et al. 2009). When forensic reports and scientific journal articles give an estimation of ancestry, it is crucial to keep in mind that responsible assignments of ancestry will be done through robust statistical testing and stated as a probability estimate. Today, we also live in a more globalized world where a skeletal individual may have been born originally to parents of two separate traditional racial categories. In contexts of great heterogeneity within populations, this definitely adds difficulty to the work of forensic scientists and anthropologists preparing results for the courtroom.