The genome is all of the genetic material for an organism. In the case of humans, this includes 46 chromosomes and mtDNA. The human genome contains approximately three billion base pairs of DNA and has regions that are both noncoding and coding. Scientists now estimate that the human genome contains 20,000–25,000 protein-coding genes, with each chromosome containing a few hundred to a few thousand genes. As our knowledge of heredity increases, researchers have begun to realize the importance of epigenetics, or changes in gene expression that do not result in a change of the underlying DNA sequence. Epigenetics research is also crucial for unraveling gene regulation, which involves complex interactions between DNA, RNA, proteins, and the environment.
Genomics
The vast majority of the human genome is noncoding, meaning there are no instructions to make a protein or RNA product in these regions. Historically, noncoding DNA was referred to as “junk DNA” because these vast segments of the genome were thought to be irrelevant and non-functional. However, continual improvement of DNA sequencing technology along with world-wide scientific collaborations and consortia have contributed to our increased understanding of how the genome functions. Through these technological advances and collaborations, we have since discovered that many of these noncoding DNA regions are involved in dynamic genetic regulatory processes.
Genomics is a diverse field of molecular biology that focuses on genomic evolution, structure and function; gene mapping; and genotyping (determining the alleles present). Evolutionary genomics determined that humans and chimpanzees share a significant portion of shared DNA sequence (about 98.8%). Given the phenotypic differences between humans and chimpanzees, having a DNA sequence difference of 1.2% seems surprising. However, a lot of genomics research is also focused on understanding how noncoding genomic regions influence how individual genes are turned “on” and “off” (i.e., regulated). Therefore, although DNA sequences are identical, regulatory differences in noncoding genetic regions (e.g., promoters) are believed to be largely responsible for the physical differences between humans and chimpanzees.
Further understanding of genomic regulatory elements can lead to new therapies and personalized treatments for a broad range of diseases. For example, targeting the regulatory region of a pathogenic gene to “turn off” its expression can prevent its otherwise harmful effects. Such molecular targeting approaches can be personalized based on an individual’s genetic makeup. Genome-wide association studies (GWAS) seek to determine genes that are linked to complex traits and diseases and typically require significant computational efforts. This is because millions of DNA sequences must be analyzed and GWAS sometimes include thousands of participants. During the beginning of the genomics field, most of the large-scale genomics studies only included North American, European, and East Asian participants and patients. Researchers are now focusing on increasing ethnic diversity in genomic studies and databases. In turn, accuracy of individual disease risk across all human populations will be improved and more rare-disease-causing alleles will be identified.
Epigenetics
All cells within your body have the same copy of DNA. For example, a brain neuron has the same DNA blueprint as does a skin cell on your arm. Although these cells have the same genetic information, they are considered specialized. The reason all cells within the body have the same DNA but different morphologies and functions is that different subsets of genes are turned “on” and “off” within the different cell types. A more precise explanation is that there is differential expression of genes among different cell types. In the case of neuronal cells, a unique subset of genes are active that allow them to grow axons to send and receive messages. This subset of genes will be inactive in non-neuronal cell types such as skin cells. Epigenetics is a branch of genetics that studies how these genes are regulated through mechanisms that do not change the underlying DNA sequence. Special Topics: Epigenetics and X Chromosome Inactivation details a well-known example of epigenetic regulation.
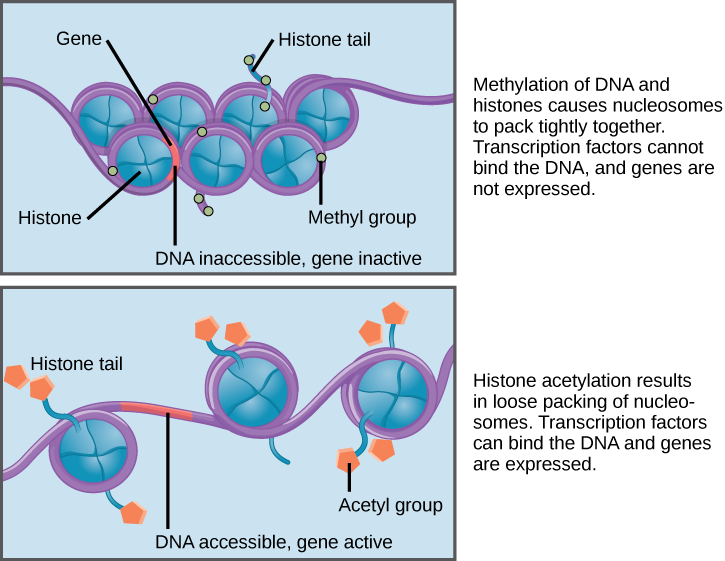
The prefix epi means “on, above, or near,” and epigenetic mechanisms such as DNA methylation and histone modifications occur on, above, or near DNA. The addition of a methyl group (—CH₃) to DNA is known as DNA methylation (Figure 3.38). DNA methylation and other modifications made to the histones around which DNA are wrapped are thought to make chromatin more compact. This DNA is inaccessible to transcription factors and RNA polymerases, thus preventing genes from being turned on (i.e., transcribed). Other histone modifications have the opposite effect by loosening chromatin, which makes genes accessible to transcription factors.
It is important to note that environmental factors can alter DNA methylation and histone modifications and also that these changes can be passed from generation to generation. For example, someone’s epigenetic profile can be altered during a stressful time (e.g., natural disasters, famine, etc.), and those regulatory changes can be inherited by the next generation. Moreover, our epigenetic expression profile changes as we age. For example, certain places in our genome become “hyper” or “hypo” methylated over time. Identical twins also have epigenetic profiles that become more different as they age. Researchers are only beginning to understand what all of these genome-wide epigenetic changes mean. Scientists have also discovered that changes in epigenetic modifications can alter gene expression in ways that contribute to diseases. It is also important to note that, unlike DNA mutations (which permanently change the nucleotide sequence), epigenetic changes can be easily reversed. A lot of research now focuses on how drugs can alter or modulate changes in DNA methylation and histone modifications to treat diseases such as cancer.
Figure \(\PageIndex{1}\): Different types of epigenetic histone tail modifications that can tighten (top) and loosen (bottom) the chromatin of DNA.
SPECIAL TOPIC: EPIGENETICS AND X CHROMOSOME INACTIVATION
Figure \(\PageIndex{2}\): A multicolored coat pattern as the result of X chromosome inactivation during development.
Mary Lyon was a British geneticist that presented a hypothesis for X chromosome inactivation (called the Lyon hypothesis) based on her work and other studies of the day. Females inherit two X chromosomes, one from each parent. Males have one functional X chromosome; however, this does not mean females have more active genes than males. During the genetic embryonic development of many female mammals, one of the X chromosomes is inactivated at random, so females have one functional X chromosome. The process of X chromosome inactivation in females occurs through epigenetic mechanisms, such as DNA methylation and histone modifications. Recent studies have analyzed the role of a long noncoding RNA called X-inactive specific transcript (XIST), which is largely responsible for the random silencing of one of the X chromosomes. The presence of two X chromosomes is the signal for XIST RNA to be expressed so that one X chromosome can be inactivated. However, some cells may have an active paternal X chromosome while other cells may have an active maternal X chromosome. This phenomenon is easily seen in calico and tortoiseshell cats (Figure 3.39). In cats, the gene that controls coat color is found on the X chromosome. During early embryo development, random inactivation of X chromosomes gives rise to populations of cells that express black or orange, which results in the unique coat patterning. Therefore, calico cats are typically always female.